
Mathematical models enter clinical trials
Mathematics is the language of physics that describes and interprets the phenomena of the natural world around us. In contrast to the often wobbly meanings of natural language, the mathematical vocabulary has defined meanings, the grammar is flawless, and the semantics are unambiguous. Thus, mathematical models are precise and accurate. They have served us well to understand the natural world, the cosmos and technology. They have enabled amazing technologies such as cars, airplanes, space flight, the telephone, TV, the internet, online communications, video games and many other commodities that we could not imagine life without.

However, there is a final frontier which mathematics struggled to get to grips with. This frontier is human biology. This was not for lack of trying. The 17th century mathematician and philosopher Rene Descartes thought of the human body as an elaborate machine that could be understood by the rules of mathematics and physics. Three hundred years later, 1944, Erwin Schroedinger in his famous little book “What is life?” inferred general properties of genes and inheritance using physical and mathematical principles. In 1952, Alan Hodgkin and Andrew Huxley devised a mathematical model that could explain the electrical processes involved in the initiation and propagation of nerve signals. They received the 1963 Nobel Prize in Physiology or Medicine for this work. In 1960, Denis Noble developed a mathematical model of the electrical processes that commands the heartbeat. However, the going became tough when mathematical physics dove into biological processes that apparently did not follow carved out physics rules such as electricity. Nevertheless, the next good stab was metabolism, which describes the pathways our cells use to produce energy and building blocks for the synthesis of important biomolecules. In the 1970s, Kacser & Burns and Heinrich & Rapoport developed metabolic control analysis, a theory that still drives the bioreactors that now produce insulin, therapeutic antibodies, vaccines and complex chemical compounds for various applications in healthcare and the bioeconomy.
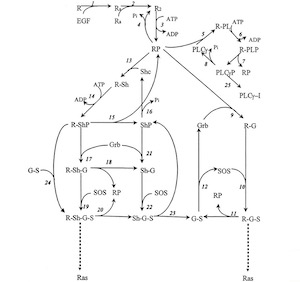
Boris Kholodenko's mathematical model of EGF receptor signaling
However, when it came to explaining information processing in biology, a process called signal transduction, we were stunned. Unlike in metabolic pathways where every worker (which is an enzyme encoded by a gene) has a defined task, signal transduction workers multitask and moonlight. They process a myriad and often ambiguous array of information to assure that a cell does the right thing at the right time and carries out the right functions 24/7 every day for your whole life. No wonder things can go wrong there. It was thought impossible to model this complexity. Using an ingenious approach, Boris Kholodenko, our SBI co-director, and his colleagues cracked the problem. They designed a kinetic, mechanistic mathematical model that could explain how the EGF receptor processes external signals1. Even better, the model could predict what happens under certain conditions, meaning we can ask “What if?” questions. What happens if the EGF concentration changes? What if we have a mutation in the EGF receptor or one of its downstream signaling elements? What if we block some of the signaling pathways using drugs? The power to ask good questions is arguably a driving force of scientific discovery. Using this type of mathematical model we now could harness this power to ask some important “How?” and “Why?” questions about cancer.
 We know that cancer is a result of these signal transduction networks becoming corrupted, but we do not really know how and why. The “why” seemed to be easy once we had figured out that some of the genetic mutations that drive cancer do so by changing the activities of signaling networks. Unfortunately, we have much less idea about the “how”. What do cancer causing mutations actually do? This is where mathematical modeling comes to the fore. It can explain mechanisms. There was one very peculiar puzzle that defied biological intuition. It was RAF inhibitors. RAF kinases are major effectors of RAS proteins that activate a downstream signaling pathway consisting of two other kinases, MEK and ERK. Mutations in RAS or RAF may be responsible for driving half or more of all human cancers including prevalent and deadly cancers such as melanoma, lung, colorectal and pancreatic cancer. More than 90 percent of pancreatic cancers have RAS mutations and >80 percent of melanomas have mutations in RAS or RAF genes. RAS is a difficult drug target. It has no pockets where a small molecule drug could wedge in to block its function. However, the solution seemed clear. We know how to make good kinase inhibitors. As RAF, MEK and ERK are kinases that transduce the cancerous growth signals and program cells to grow relentlessly, extremely potent RAF and MEK inhibitors were developed. We all expected that they would block oncogenic signaling by mutated RAS. Surprisingly, they did not. In the case of RAF inhibitors, they even enhanced the activation of MEK and ERK. This paradoxical pathway activation is a major reason why patients are resistant or become resistant to these drugs. However, these drugs worked in melanoma patients who had suffered a RAFmutation. Here we were in 2010. We had probably the best kinase inhibitors ever made by pharma, and they behaved strangely. We really could not understand why they worked in melanoma patients with RAF mutations but not in melanoma patients with RAS mutations, and in fact, in no other cancers with RAS mutations. Hundreds of excellent research papers were produced in the search of an explanation, but it took a mathematical model to come up with a universal rationale for what was happening. The biological UFO sighting started to make sense and became an explainable twist of Nature.
We know that cancer is a result of these signal transduction networks becoming corrupted, but we do not really know how and why. The “why” seemed to be easy once we had figured out that some of the genetic mutations that drive cancer do so by changing the activities of signaling networks. Unfortunately, we have much less idea about the “how”. What do cancer causing mutations actually do? This is where mathematical modeling comes to the fore. It can explain mechanisms. There was one very peculiar puzzle that defied biological intuition. It was RAF inhibitors. RAF kinases are major effectors of RAS proteins that activate a downstream signaling pathway consisting of two other kinases, MEK and ERK. Mutations in RAS or RAF may be responsible for driving half or more of all human cancers including prevalent and deadly cancers such as melanoma, lung, colorectal and pancreatic cancer. More than 90 percent of pancreatic cancers have RAS mutations and >80 percent of melanomas have mutations in RAS or RAF genes. RAS is a difficult drug target. It has no pockets where a small molecule drug could wedge in to block its function. However, the solution seemed clear. We know how to make good kinase inhibitors. As RAF, MEK and ERK are kinases that transduce the cancerous growth signals and program cells to grow relentlessly, extremely potent RAF and MEK inhibitors were developed. We all expected that they would block oncogenic signaling by mutated RAS. Surprisingly, they did not. In the case of RAF inhibitors, they even enhanced the activation of MEK and ERK. This paradoxical pathway activation is a major reason why patients are resistant or become resistant to these drugs. However, these drugs worked in melanoma patients who had suffered a RAFmutation. Here we were in 2010. We had probably the best kinase inhibitors ever made by pharma, and they behaved strangely. We really could not understand why they worked in melanoma patients with RAF mutations but not in melanoma patients with RAS mutations, and in fact, in no other cancers with RAS mutations. Hundreds of excellent research papers were produced in the search of an explanation, but it took a mathematical model to come up with a universal rationale for what was happening. The biological UFO sighting started to make sense and became an explainable twist of Nature.
The breakthrough was a mathematical model that could not only look at the bare signaling events, but could also consider context2, such as the type of mutations present in a cancer cell, the networks the mutant proteins are embedded in, the protein interactions they participate in, their modifications of proteins, their structure and their ability to bind drugs. We called this “Next Generation Dynamic Modelling”. Developing this model was a major joint effort between SBI and the groups of Bill Hlavacek and Richard Posner in the USA, and Edina Rosta in the UK. However, the name lived up to the claim. We were able to explain how resistance to RAF inhibitors occurs on a network level down to the molecular details of protein structure and thermodynamics of drug binding. This was no small feat, and although the model only looks at a small pathway, it was unprecedented in its depth and predictive power. We had a groundbreaking model that was experimentally verified.

Where do we go from here? Getting an exciting research finding into clinical application is not easy. It is slow, tedious, and fraught with regulations and approvals that make a researcher’s head spin and motivation sink. A mathematical model going to clinical trial was particularly unheard of. Time to call it a day and quit? Fortunately, our model’s potential solution for tackling mutant RAS driven cancers caught the eyes of a clinical team around Prof Dan Von Hoff and Dr Erkut Borazanci at the HonorHealth Research Institute of Scottsdale, Arizona, USA. They had lost a young, valued colleague, Dr Nate Nieto, to pancreatic cancer and were keen on trying a new approach to this deadly cancer. Pancreatic cancer is driven by KRAS gene mutations, which are currently not druggable. Our model, however, predicted that a certain combination of RAF inhibitors could block certain types of KRAS mutants. This is how the clinical trial was designed. It will recruit pancreatic cancer patients who have a certain type of KRAS mutation and treat them with a combination of RAF inhibitors which we found to synergistically inhibit signaling by this particular KRAS mutation. The clinical trial is entitled “Phase II Trial of Vemurafenib and Sorafenib in Pancreatic Cancer” (https://clinicaltrials.gov/ct2/show/NCT05068752 ), and has started recruiting patients.
To the best of our knowledge, this is the very first clinical trial based on a dynamic, mechanistic mathematical model. This is a groundbreaking change of paradigm in many ways. Most importantly, clinical trials are designed to test population responses. This trial selects patients based on expected personal treatment responses. In fact, we could make personal computational models, so-called Digital Twins, for each individual patient to select the best possible treatment. Current clinical trial protocols do not accommodate that. To change this, we need successful examples showing that personalized therapies can be better than the therapies based on population averages. The Nate Nieto trial may well be a first and all-important step to provide personalized therapies with the help of computer models.
References
1. Kholodenko, B. N., O. V. Demin, G. Moehren, and J. B. Hoek. Quantification of short term signaling by the epidermal growth factor receptor. J Biol Chem 274:30169-81, 1999.
2. Rukhlenko, O. S., F. Khorsand, A. Krstic, J. Rozanc, L. G. Alexopoulos, N. Rauch, K. E. Erickson, W. S. Hlavacek, R. G. Posner, S. Gómez-Coca, E. Rosta, C. Fitzgibbon, D. Matallanas, J. Rauch, W. Kolch, and B. N. Kholodenko. Dissecting RAF Inhibitor Resistance by Structure-based Modeling Reveals Ways to Overcome Oncogenic RAS Signaling. Cell Syst 7:161-179.e14, 2018.

